
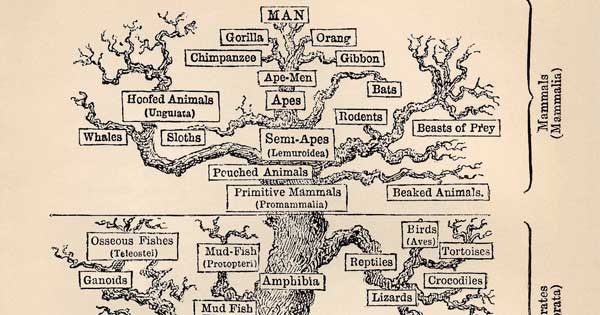
Uno de los pilares centrales del modelo evolutivo estándar es la creencia de que todas las especies vivientes evolucionaron a partir de un antepasado común a través de un árbol de la vida que se fue desplegando gradualmente. Como resultado, se cree que el patrón de similitudes y diferencias en las especies actuales encaja en un patrón similar a un árbol o una jerarquía anidada en la que los puntos de ramificación corresponden a la aparición de nuevos rasgos biológicos. Por ejemplo, todos los mamíferos comparten ciertas características, como la producción de leche, ya que su ancestro común más reciente desarrolló por primera vez esos rasgos y los rasgos que se transmiten a través de cada rama evolutiva del árbol. Este icónico árbol de la vida ha sido presentado al público como una de las pruebas más sólidas para el desarrollo de la vida que avanza completamente a través de procesos naturales no dirigidos. Sin embargo, muchos aspectos de esta historia han sido contradichos por grandes descubrimientos en las últimas décadas.
De importancia primordial, los primeros representantes de la mayoría de los principales grupos de organismos aparecieron repentinamente en el registro fósil sin secuencias identificables de intermedios que condujeran de nuevo a un ancestro común con otros grupos. Igualmente problemático, el patrón de rasgos físicos y secuencias moleculares en especies en toda la naturaleza no implica un árbol evolutivo consistente. Por ejemplo, los ojos de los humanos y los pulpos son bastante similares, pero se cree que los dos grupos solo están relacionados lejanamente. Resaltando este desafío, un artículo más antiguo analizó en varios estudios los porcentajes de características para grupos de especies dados que encajan de manera consistente con los cladogramas mejor construidos (aproximaciones a un árbol evolutivo). Estos porcentajes conocidos como «índices de consistencia» se trazaron en el mismo gráfico que los derivados de datos generados aleatoriamente, y los índices se ajustaron para eliminar el efecto del ruido aleatorio. El promedio para los índices ajustados residió en algún lugar alrededor de 0.35. Los intentos más recientes de construir cladogramas para varios grupos no han sido mejores, como con Euarchonta (grupo que incluye a los primates) y Therapsida (ancestros propuestos a los mamíferos). En otras palabras, aproximadamente dos tercios de todos los datos no se ajustan al modelo de antepasado común.
Resultados decepcionantes, mecanismos especiales
Estos decepcionantes resultados han requerido que los evolucionistas ideen varios mecanismos ad hoc para explicar las incoherencias ubicuas. Los ejemplos incluyen transferencia génica lateral (TGL), pérdida diferencial de genes y evolución convergente. Sin embargo, la apelación a la Transferencia Génica Lateral ha sido seriamente cuestionada. Y la afirmación de que las adaptaciones complejas pueden aparecer de manera independiente múltiples veces (evolución convergente) colapsa en un examen minucioso debido a la imposibilidad de su aparición a través de procesos no dirigidos, incluso una vez.
Por ejemplo, se cree que los ojos con lentes han evolucionado independientemente varias veces, pero todos los escenarios se enfrentan a barreras insuperables en términos de presiones selectivas opuestas y escalas temporales requeridas. Aún más sorprendente, la supuesta evolución convergente de la ecolocalización en murciélagos y delfines involucró las mismas modificaciones de secuencia en más de 200 regiones de su ADN. Sin embargo, el tiempo disponible para que un mamífero terrestre evolucione en un animal marino totalmente acuático es insuficiente para adquirir incluso dos nuevas mutaciones coordinadas, y la transformación a mamíferos acuáticos requirió muchas modificaciones adicionales. A pesar de estos desafíos, los evolucionistas han mantenido que el ancestro común sigue siendo la mejor explicación para los datos, ya que en cierto modo se ajusta a un patrón similar a un árbol. Esa postura ahora se enfrenta a un desafío formidable de un artículo reciente en la revista BIO-Complexity, de Winston Ewert. Él persuasivamente presenta un nuevo marco para explicar mejor el patrón de la naturaleza.
Un nuevo marco
El modelo de Ewert interpreta el patrón de similitudes en diferentes grupos de especies compatibles con lo que se conoce en informática como gráficos de dependencia. Específicamente, los programadores no suelen escribir nuevos programas completamente desde cero. En su lugar, reutilizan los módulos estándar. Un ejemplo es la solicitud del módulo de JavaScript, que descarga archivos de Internet. Los módulos generalmente acceden («solicitan») a otros módulos, formando una red de ramificación de relaciones de dependencia (ver Figura 1).
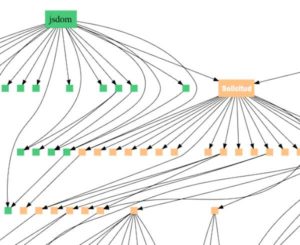
Figura 1: Gráfico de dependencia que ilustra el módulo jsdom que accede a la solicitud del módulo que accede a otros módulos. También se accede a la solicitud por otros módulos de nivel superior.
Los gráficos de dependencia a menudo incluyen una jerarquía anidada como parte de su estructura, pero también contienen relaciones adicionales que se extienden más allá de un árbol simple. La diferencia puede verse al comparar el árbol de vida de mamífero estándar (Figura 2) basado en el antepasado común con un gráfico de dependencia derivado de la misma especie (Figura 3).
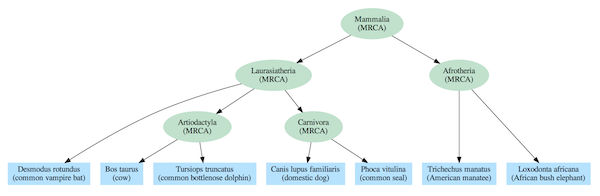
Figura 2: Árbol de la vida para especies seleccionadas de mamíferos. Cada conjunto de especies en un nivel dado en la jerarquía anidada tiene solo un ancestro común más reciente.
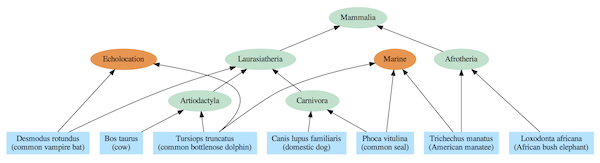
Figura 3: gráfico de dependencia para especies de mamíferos seleccionadas. Una sola especie puede depender de múltiples módulos.
El modelo de árbol evolutivo requiere, al menos para la vida compleja, que cada especie se vincule con un antepasado común más reciente (ACMR) con otras especies del mismo grupo (clado). Por ejemplo, los elefantes y los manatíes son miembros del clado Afrotheria, por lo que se los representa con un ACMR que fue el primer representante de ese clado. Y, cada ACMR se vincula a un solo ACMR para todos los miembros de clados en niveles superiores en la jerarquía. Por ejemplo, los ACMR de los clados Artiodactyla y Carnivora comparten un único ACMR con todos los miembros del clado de Laurasiatheria. Y todas las especies de mamíferos se vinculan a un solo ACMR que es el tronco del árbol de la vida de Mammalia.
El gráfico de dependencia correspondiente incluye la misma jerarquía anidada que el supuesto árbol evolutivo, pero las relaciones se interpretan no en términos de ACMR sino en términos de módulos compartidos. Por ejemplo, los elefantes y los manatíes usan el módulo Afrotheria y todos los mamíferos usan el módulo Mammalia. Sin embargo, en marcado contraste con el modelo de antepasado común, los manatíes, las focas y los delfines utilizan el módulo marino que es incongruente con el árbol evolutivo. Del mismo modo, los murciélagos y los delfines usan el módulo de ecolocalización, mientras que en el modelo de antepasado común no están estrechamente relacionados.
Comparando dos modelos
Ewert comparó el poder predictivo del gráfico de dependencia y los modelos de antepasados comunes mediante el análisis de la distribución de familias de genes en diferentes colecciones de especies tomadas de nueve bases de datos diferentes. Para cada base de datos, se identificó un conjunto de genes utilizados por múltiples especies como un módulo del que dependen esas especies. Y un conjunto de genes contenidos en múltiples módulos más grandes se identificó como un módulo distinto del que dependen los módulos más grandes. Se usó una rutina de optimización para construir una aproximación al mejor gráfico de dependencia, y ese gráfico se comparó con el árbol de la vida presentado por la jerarquía de la base de datos del NCBI (National Center for Biotechnology Information Search database). El mejor ajuste para los datos se determinó luego entre las dos representaciones utilizando la selección del modelo bayesiano.
El modelo de gráfico de dependencia hace varias predicciones que están en oposición directa al modelo de antepasado común:
- Los datos biológicos deberían ajustarse a un gráfico de dependencia mejor que un árbol.
- Los datos producidos por un proceso dominado por una descendencia o ramificación común deberían ajustarse mejor a un árbol que un gráfico de dependencia.
- Los gráficos inferidos para datos biológicos deberían contener muchos más módulos no taxonómicos con muchos más genes que los gráficos de dependencia inferidos a partir de datos que se sabe que fueron producidos por descendencia común.
- El software debe ajustarse a un gráfico de dependencia mejor que a un árbol, pero un árbol es mejor que un modelo nulo. Un modelo nulo corresponde a ningún patrón existente para la reutilización de las familias de genes en todas las especies.
El análisis de Ewert validó todas estas predicciones con una alta confianza estadística para todas las bases de datos. Por lo tanto, este estudio inicial sugiere que el modelo de gráfico de dependencia supera en gran medida el modelo del árbol con antepasado común, para comprender el patrón de la naturaleza.
Como consecuencia, todos los supuestos árboles evolutivos y secuencias se vuelven altamente sospechosos, incluidos iconos como las series de ballenas y humanos. Porque se basan en similitudes de rasgos entre especies, y las similitudes son un indicador poco confiable de ancestros comunes como lo implican los índices de consistencia ajustados típicamente bajos de los árboles. En cambio, las similitudes parecen ser el resultado de un diseñador que reutiliza módulos de diseño en diferentes especies para alcanzar objetivos comunes.
El artículo de Ewert representa solo el primer paso para evaluar y desarrollar su marco. Aún así, la importancia de esta investigación no puede ser exagerada. El modelo de gráficos de dependencia explica por qué los subconjuntos de datos biológicos se ajustan groseramente a un patrón de árbol y por qué gran parte de los datos son incongruentes. También hace predicciones claras sobre los resultados de futuros estudios sobre la distribución entre especies de ambos rasgos físicos y similitudes en datos moleculares. Finalmente, debe conducir a un programa de investigación robusto e innovador basado en el marco del diseño inteligente.
Artículo publicado originalmente en inglés por Brian Miller Ph.D.
Imagen: Un árbol de la vida tradicional, a través de Wikimedia Commons.



{kind=link}